Benchmark Tasks
Designing Organic Photovoltaics
Our first set of benchmark objectives consists of six individual tasks that are inspired by the design of organic photovoltaics (OPVs). The development of organic solar cells (OSCs) is of wide interest because of their potential to replace the currently predominating inorganic devices and to expand upon their application domain.
The code accepts a proposed structure as SMILES string, generates initial Cartesian coordinates with Open Babel, and performs conformer search and geometry optimization with crest and xtb, respectively. Finally, a single point calculation at the GFN2-xTB level of theory provides the properties of interest, in particular the HOMO and LUMO energies, the HOMO-LUMO gap and the molecular dipole moment. The power conversion efficiency (PCE) is computed from these simulated properties based on the Scharber model.
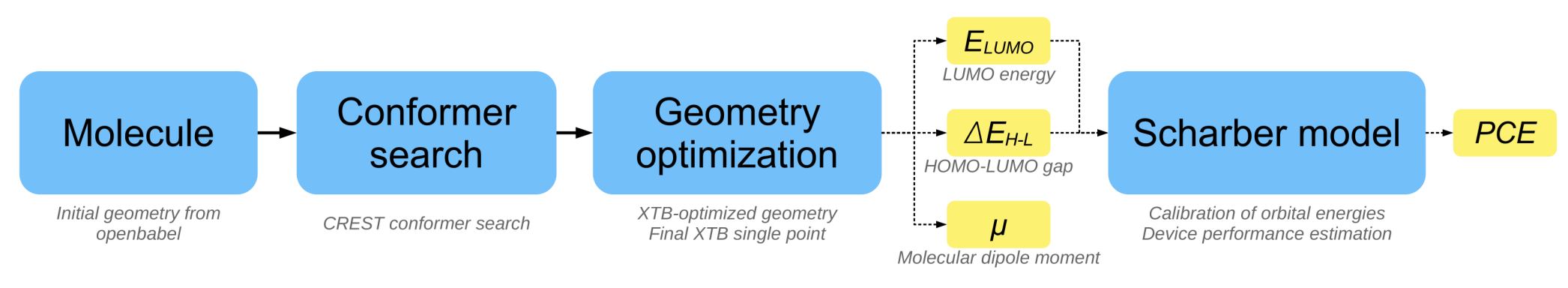
Designing organic photovoltaics.
Example
import pandas as pd
data = pd.read_csv('./datasets/hce.csv') # or ./dataset/unbiased_hce.csv
smiles = data['smiles'].tolist()
smi = smiles[0]
## use full xtb calculation in hce module
from tartarus import pce
pce_pcbm_sas, pce_pcdtbt_sas = pce.get_properties(smi)
Designing Organic Emitters
The next set of benchmarks is inspired by the design of purely organic emissive materials for organic lightemitting diodes (OLEDs), which received significant attention in recent years after the discovery of thermally activated delayed fluorescence (TADF) in the field.
The code accepts a proposed structure as SMILES string and starts with conformer search for the molecule. Subsequent geometry optimization via xtb provides the structure used in the final TD-DFT single point calculation with pyscf. From the output, the singlet-triplet gap, oscillator strength and vertical excitation energy is extracted and returned.
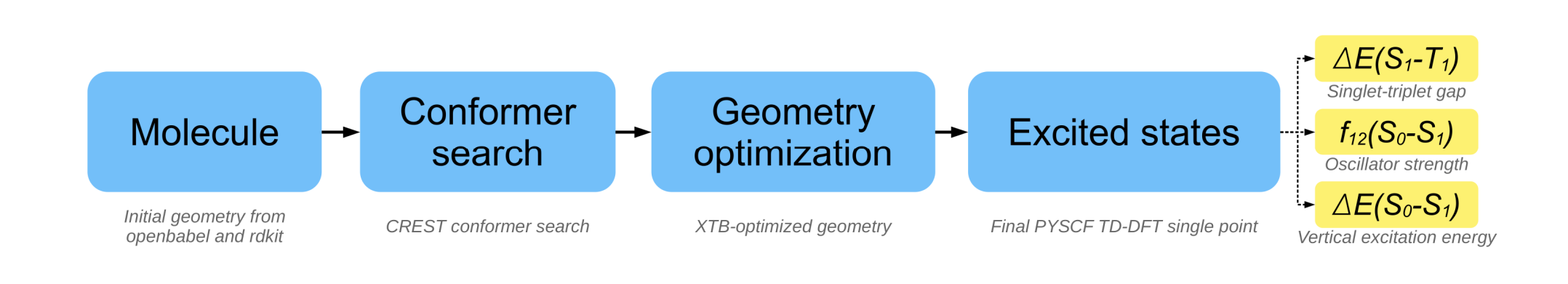
Designing organic emitters
Example
import pandas as pd
data = pd.read_csv('./datasets/gdb13.csv')
smiles = data['smiles'].tolist()
smi = smiles[0]
## use full xtb calculation in hce module
from tartarus import tadf
st, osc, combined = tadf.get_properties(smi)
Design of Drug Molecule
Next, we also wanted to include molecular design objectives that are relevant for medicinal chemistry in TARTARUS. In recent years, deep generative models have experienced a strong increase in popularity and adoption for drug design as they promise to accelerate discovery campaigns leading to significant cost reductions, and several success stories were reported on in the literature.
The code accepts a proposed structure as SMILES string, generates initial Cartesian coordinates with Open Babel and then samples docking poses in the predefined binding sites using smina. The lowest docking score of all the sampled poses is returned as target property.
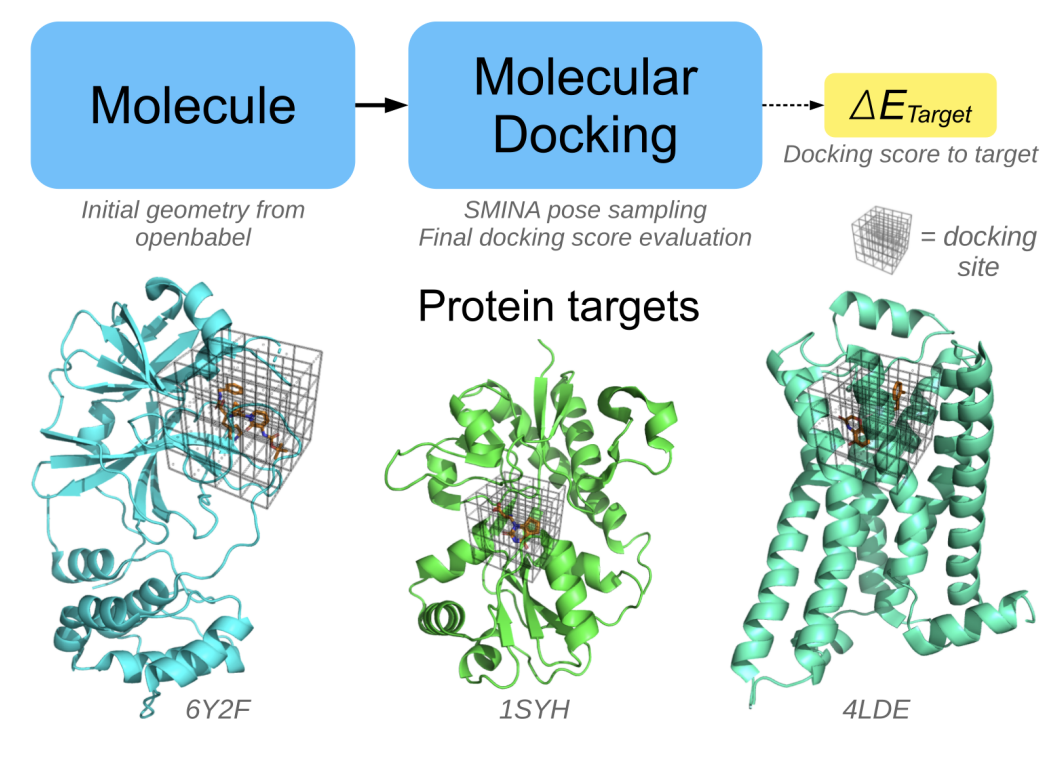
Example
import pandas as pd
data = pd.read_csv('./datasets/docking.csv')
smiles = data['smiles'].tolist()
smi = smiles[0]
## Design of Protein Ligands
from tartarus import docking
score_1syh = docking.perform_calc_single(smi, '1syh', docking_program='qvina')
Design of Chemical Reaction Substrates
Developing new chemical reactions and finding new catalysts for existing ones are important goals to drive innovations in drug and material discovery, and move towards more sustainable chemical production. Whereas, classically, the optimization of reaction parameters was largely dominated by experimental work, in recent years, the significant increase in computing power and the continuous improvement of computer algorithms enabled molecular simulations to play an increasingly important part.
The code accepts a proposed structure as SMILES string and starts with optimizations and conformer searches for the reactants and products. The SEAM optimization is used to generate a guess transition state, which is then subjected to constrained conformational sampling. From the final structures, the reaction energy and approximated SEAM activation energy are extracted and returned.

Design of Chemical Reaction Substrates
Example
import pandas as pd
data = pd.read_csv('./datasets/reactivity.csv')
smiles = data['smiles'].tolist()
smi = smiles[0]
## calculating binding affinity for each protein
from tartarus import reactivity
Ea, Er, sum_Ea_Er, diff_Ea_Er = reactivity.get_properties(smi)
Datasets
See below for a summary of the contents of the datasets included in TARTARUS. The datasets are available in the datasets folder.
Task |
Dataset name |
# of smiles |
Columns in file |
|||||
|---|---|---|---|---|---|---|---|---|
Designing OPV |
hce.csv |
24,953 |
Dipole moment (↑) |
HOMO-LUMO gap (↑) |
LUMO (↓) |
Combined objective (↑) |
PCE<sub>PCBM</sub> -SAS (↑) |
PCE<sub>PCDTBT</sub> -SAS (↑) |
Designing emitters |
gdb13.csv |
403,947 |
Singlet-triplet gap (↓) |
Oscillator strength (↑) |
Multi-objective (↑) |
|||
Designing drugs |
docking.csv |
152,296 |
1SYH (↓) |
6Y2F (↓) |
4LDE (↓) |
|||
Designing chemical reaction substrates |
reactivity.csv |
60,828 |
Activation energy ΔE<sup>‡</sup> (↓) |
Reaction energy ΔE<sub>r</sub> (↓) |
ΔE<sup>‡</sup> + ΔE<sub>r</sub> (↓) |
|